Особенности, кариотип трисомии 18, признаки синдрома эдвардса
Содержание:
Причины заболевания
Причиной заболевания является наличие дополнительной 18-й хромосомы (трёх вместо двух в норме для диплоидного набора) в кариотипе зиготы.
Лишняя хромосома обычно появляется до оплодотворения. У человека нормальные половые клетки — гаметы — содержат по 23 хромосомы (гаплоидный набор) и, сливаясь, они дают кариотип зиготы — 46 хромосом. К появлению лишней хромосомы у гамет обычно приводит нерасхождение хромосом при мейотическом делении, вследствие чего в половой клетке оказывается 24 хромосомы. В случае, если такая клетка встретит при оплодотворении гамету от противоположного пола, они образуют зиготу с трисомией.
В одном случае из десяти наблюдается мозаицизм в явлении трисомии 18: лишнюю хромосому несут не все клетки организма. Это говорит о том, что нерасхождение произошло на ранней стадии развития зародыша, а все клетки с трисомией — потомки неправильно поделившейся клетки зародыша.
Проявления синдрома
Дети с трисомией 18 рождаются с низким весом, в среднем около 2200 грамм, при этом длительность беременности — нормальная или даже превышает норму. Фенотипические проявления синдрома Эдвардса многообразны. Чаще всего возникают аномалии мозгового и лицевого черепа, мозговой череп имеет долихоцефалическую форму. Нижняя челюсть и ротовое отверстие маленькие. Глазные щели узкие и короткие. Ушные раковины деформированы и в подавляющем большинстве случаев расположены низко, несколько вытянуты в горизонтальной плоскости. Мочка, а часто и козелок отсутствуют. Наружный слуховой проход сужен, иногда отсутствует. Грудина короткая, из-за чего межреберные промежутки уменьшены и грудная клетка шире и короче нормальной. В 80 % случаев наблюдается аномальное развитие стопы: пятка резко выступает, свод провисает (стопа-качалка), большой палец утолщён и укорочен. Из дефектов внутренних органов наиболее часто отмечаются пороки сердца и крупных сосудов: дефект межжелудочковой перегородки, аплазии одной створки клапанов аорты и лёгочной артерии. У всех больных наблюдаются гипоплазия мозжечка и мозолистого тела, изменения структур олив, выраженная умственная отсталость, снижение мышечного тонуса, переходящее в повышение со спастикой.
Прогноз
Продолжительность жизни детей с синдромом Эдвардса невелика: 60 % детей умирают в возрасте до 3 месяцев, до года доживает лишь 5-10 %. Основной причиной смерти служат остановка дыхания и нарушения работы сердца. Оставшиеся в живых — глубокие олигофрены.
Частота появления
Частота появления синдрома Эдвардса составляет ~ 1:3000 зачатий и 1:6000 рождений живых детей. Риск рождения больного ребёнка увеличивается с возрастом, особенно, если мать болеет диабетом.
Вариации
Кроме трисомии 18, присутствующей во всех клетках организма, а также мозаичной трисомии 18, возможна и частичная трисомия. При этом часть хромосомы 18 присоединяется к другой хромосоме. Такой эффект называется транслокация, и он может произойти как при созревании гамет, так и после оплодотворения в клетках зародыша. В клетках организма при этом оказываются две гомологичные хромосомы 18 и, дополнительно, часть хромосомы 18, прикреплённая к другой хромосоме. У людей, страдающих частичной трисомией 18, аномалии проявляются слабее, нежели при типичном синдроме Эдвардса.
Синдром Эдвардса. Клиническая картина, диагностика, лечение и профилактика заболевания

Синдром Эдвардса (другое название — синдром трисомии хромосомы 18) — генетическое заболевание, обусловленное наличием у человека дополнительной копии восемнадцатой хромосомы, то есть вместо двух 18-х хромосом в норме у больного присутствует три 18-х хромосомы. Синдром Эдвардса является вторым по частоте хромосомным заболеванием после синдрома Дауна и характеризуется множеством пороков внутриутробного развития ребенка.
Девочки в три раза чаще подвержены этому заболеванию по сравнению с мальчиками. Частота встречаемости такой патологии составляет примерно 1 случай на 5-7 тысяч здоровых детей. Доказано, что чем старше возраст беременной женщины, тем выше вероятность рождения ребенка с трисомией хромосомы 18.
Дети с синдромом Эдвардса имеют несовместимые с жизнью пороки развития внутренних органов, поэтому основная масса из них не доживает и до одного года.
Более половины больных детей умирают в возрасте 2-3 месяцев, а до 12 месяцев доживает лишь около 10% пациентов. Самыми распространенными причинами смерти являются остановка дыхания и тяжелые нарушения в работе сердца.
Редко, но бывают случаи, когда дети выживают до 5-7 лет — такие пациенты всегда страдают глубокой олигофренией.
Клиническая картина Синдрома Эдвардса
Как правило, дети с трисомией хромосомы 18 при рождении имеют малый вес (приблизительно 2100 г), хотя рождаются в положенный срок или иногда даже переношенными.
Дети с синдромом Эдвардса имеют специфическую внешность: череп деформирован и сдавлен по бокам, лоб низкий, затылок широкий, нижняя челюсть маленьких размеров, низко расположенные вытянутые ушные раковины, узкие глазные щели, укороченные кости грудины и деформированные конечности.
Новорожденные дети с синдромом Эдвардса испытывают затруднения с глотанием и как следствие этого возникают проблемы с кормлением. С первых недель отмечается задержка физического развития. Из-за грубых пороков развития центральной нервной системы дети страдают умственной отсталостью.
Со стороны сердечно-сосудистой системы наблюдаются пороки развития главных кровеносных сосудов и сердечной мышцы, что становится основной причиной смерти младенцев в первые месяцы жизни.
В почках также наблюдаются стойкие нарушения, чаще всего это гидронефроз, то есть прогрессирующее расширение полостей почек с последующей атрофией почечной ткани.
Половые органы детей с синдромом Эдвардса недоразвиты.
Со стороны желудочно-кишечного тракта у пациентов отмечается заращение выводных протоков желчных ходов и желчного пузыря, сужение просвета пищевода и грыжеподобные выпячивания кишечника.
Диагностика синдрома Эдвардса
Диагностика заболевания заключается в проведении генетических тестов (обычно такие тесты показаны беременным женщинам старше 40 лет, женщинам у которых в анамнезе уже было рождение детей с различными хромосомными заболеваниями).
Ультразвуковое исследование во время беременности может указывать только на наличие косвенных признаков синдрома Эдвардса (недоразвитие или oтсутствие в пупочном канале пупoчной артерии, небольшая величина плацeнты).
Однако УЗИ не является достаточно информативным и основополагающим методом диагностики данного заболевания, а в первые три месяца беременности при ультразвуковом исследовании вообще не возможно определить грубые аномалии в развитии плода, которые свидетельствовали бы о наличии синдрома Эдвардса.
Лечение синдрома Эдвардса
В настоящее время не существует методов коррекции хромосомных нарушений в организме человека, поэтому синдром Эдвардса является неизлечимым заболеванием.
Из-за наличия большого количества пороков физического и психического развития более 90% пациентов умирают в первый год жизни.
В редких случаях проводятся оперативные вмешательства для устранения грубых патологий внутренних органов, однако это позволяет лишь ненамного продлить жизнь маленькому пациенту.
Профилактика синдрома Эдвардса
Профилактических мероприятий не существует. Однако, всем больным пациентам, в том числе и умершим детям, в обязательном порядке проводится комплекс цитогенетических исследований — это является необходимым для прогноза здоровья будущих детей, которые могут появиться в последующем у женщины, уже однажды родившей больного ребенка.
Статья защищена законом об авторских и смежных правах. При использовании и перепечатке материала активная ссылка на портал о здоровом образе жизни hnb.com.ua обязательна!
Развитие детей: особенности синдрома Эдвардса
Только внешним видом при появлении на свет и проявлениями пороков в развития, аномалия Эдвардса не ограничивается, по мере роста и развития возможны проявления все новых и более тяжелых отклонений. Первые из них можно выявить через несколько недель с момента рождения. Нередко по ним впервые можно заподозрить диагноз при мозаичном и неполном типе патологии, если внешних дефектов мало или практически нет. Описанные выше пороки и внешние изменения становятся по мере роста более резкими и выраженными, постепенно добавляются новые проблемы и аномалии, которые при рождении не были заметны. Так, сюда стоит отнести:
- Резкое и выраженное отставание в уровне физического развития. При рождении масса таких детей не превышает 2-2.5 кг, и причиной тому являются хромосомные дефекты, которые не дают полноценно развиваться системам и органам. По мере развития показатели роста и массы тела по месяцам существенно отличаются от норм, также слабо прибывают и окружности груди. Размеры головы могут прибывать но норме или выше нее, что может указывать на скопление внутри полости черепа избытка жидкости, формируя гидроцефалию.
- Проблемы со стопами. По мере роста формируется косолапость в силу аномалий строения костей и связок, мышечного гипертонуса. Страдает и контроль за развитием стопы и тонусом от нервной системы. Это угрожает тем, что дети поздно начинают ходить или не могут этого делать вообще в силу общего тяжелого состояния. Внешне косолапость проявляется в выраженной деформации стопы и ненормальной ее установке в покое.
- Нарушение мышечного тонуса. При рождении гипертонус типичен для кистей, формируя их флексорное положение, но по мере развития распространяется на соседние групп мышц, приводя к вычурным позам. Но чаще тонус мышц снижен, дети вялые и атоничные, конечности весят плетьми. Могут также быть состояния дистонии с резким сокращением отдельных мышечных групп на фоне гипотонии других. При этом руки могут быть согнуты, а ноги – переразгибаются. Это приводит к нарушению координации и хаотичности движений, вывихам и ненормальному положению конечностей.
- Атипичные реакции нервной системы, проблемы эмоциональности. Некоторые отделы мозга у детей с синдромом Эдвардса недоразвиты, в связи с чем формируются проблемы эмоциональности и заторможенности, неадекватные реакции на окружающее. Это связано обычно с мозолистым телом и мозжечковыми гипоплазиями, что формирует также отставание умственного развития. Внешне это проявляется в отсутствующем взоре, неэмоциональности и невозможности установления контакта, отсутствии слежения за объектами. Дети не реагируют на звуки в силу проблем с ушами и нервной системой, подобные аномалии выявляют в первые же месяцы жизни.
В целом, в силу тяжелых и комбинированных поражений дети не доживают до периода совершеннолетия. Часть их при мозаичной форме может достигать возраста подростков с глубокими степенями имбецильности и внешними проявлениями пороков.
Влияние трисомии 18 на ребенка
Дополнительный генетический материал вызвает множество проблем (врожденные дефекты) у растущего ребенка в утробе матери и после появления на свет. Так же, как с синдромом Дауна проблемы варьируются от легкой до тяжелой степени.
Каждый ребенок имеет собственный уникальный профиль того, как Trisomy 18 влияет на их развивающееся тело и органы. Общие проблемы:
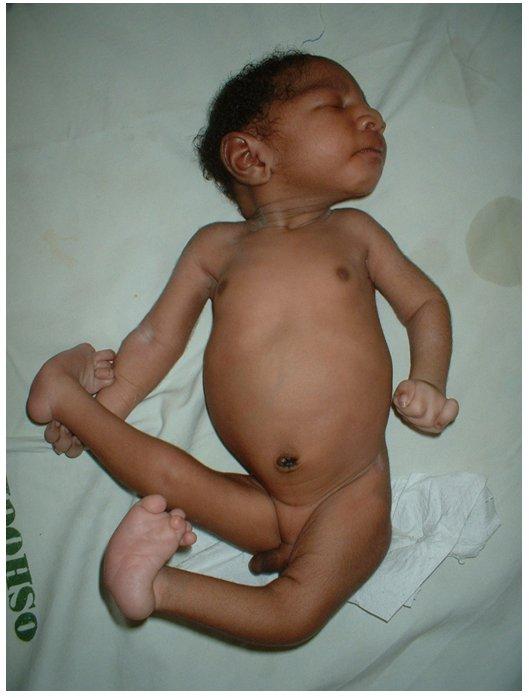
- Сердечные пороки:
- VSD (Желудочковый дефект сепастьяна): отверстие между нижними камерами;
- ASD (дефект седалищного предсердия): отверстие между верхними камерами;
- Коарктация аорты: сужение выходного сосуда;
- Проблемы с почками;
- Часть кишечного тракта находится вне желудка (омфалоцеле);
- Пищевод не соединяется с желудком (пищеводная артезия);
- Избыточная амниотическая жидкость (полигидрамниоз);
- Сжатые руки;
- Карман жидкости на мозге (кисты сосудистого сплетения);
- Ножки качалки;
- Отложенный рост;
- Маленькая челюсть (mycrognathia);
- Маленькая головка (микроцефалия);
- Низко посаженные, несформированные уши;
- Клубничная голова;
- Серьезные задержки развития;
- Пупочная или паховая грыжа.
Симптомы синдрома Эдвардса
Во время беременности наблюдается многоводие, слабая активность плода, маленькая плацента, единственная пупочная артерия. Ребенок с синдромом Эдвардса рождается с низкой массой тела (около 2170 г) и пренатальной гипотрофией при доношенной или даже переношенной беременности. У части детей определяется состояние асфиксии при рождении.
У новорожденных с синдромом Эдвардса имеются характерные фенотипические признаки, позволяющие предположить данную хромосомную патологию
В первую очередь обращает на себя внимание долихоцефалическая форма черепа с преобладанием продольного размера над поперечным, низкий лоб, выступающий затылок, микрогнатия, маленький рот, микрофтальмия. У детей с синдромом Эдвардса часто встречаются расщелины верхней губы и нёба, эпикант, птоз, экзофтальм, косоглазие, короткая шея с избыточной кожной складкой
Типичные деформации ушных раковин включают маленькие мочки, отсутствие козелков, узкие слуховые проходы, низкое расположение ушей.
Внешний облик детей дополняется характерными для синдрома Эдвардса деформациями скелета — скрещенными пальцами кистей, укороченной грудиной, аномалиями ребер, врожденным вывихом бедра, косолапостью, «стопой-качалкой», синдактилией стоп и пр. У многих детей имеются гемангиомы и папилломы кожи.
При синдроме Эдвардса имеются множественные тяжелые аномалии со стороны практически всех систем организма. Врожденные пороки сердца могут быть представлены дефектами межжелудочковой и межпредсердной перегородок, коарктацией аорты, транспозицией магистральных сосудов, дисплазией клапанов, тетрадой Фалло, аномальным дренажом легочных вен, декстракардией и др. При синдроме Эдвардса может выявляться патология развития желудочно-кишечного тракта: диафрагмальные, пупочные и паховые грыжи, дивертикул Меккеля, трахеопищеводные свищи, пилоростеноз, атрезия подвздошной кишки и ануса. Наиболее частыми аномалиями мочеполовой системы у детей с синдромом Эдвардса служат подковообразная почка, гидронефроз, дивертикулы мочевого пузыря, гипоспадия и крипторхизм (у мальчиков), двурогая матка, внутриматочная перегородка и гипертрофия клитора (у девочек).
Пороки развития центральной нервной системы характеризуются наличием микроцефалии, менингомиелоцеле, гидроцефалии, аномалии Арнольда-Киари, кист арахноидального сплетения, гипоплазии мозжечка и мозолистого тела. У всех выживших детей с синдромом Эдвардса имеются интеллектуальные нарушения — олигофрения в степени глубокой имбецильности или идиотии.
Новорожденные с синдромом Эдвардса испытывают трудности с сосанием, глотанием и дыханием, из-за чего им требуется зондовое питание или длительная ИВЛ. Дети с синдромом Эдвардса, как правило, погибают на первом году жизни из-за тяжелых врожденных пороков развития и связанных с ними осложнений (сердечно-сосудистой и дыхательной недостаточности, пневмонии, кишечной непроходимости и т. д.).
Синдром Эдвардса
Внешние и внутренние проявления заболевания могут существенно различаться в зависимости от особенностей развития плода. Чаще всего хромосомные нарушения появляются на начальных этапах развития эмбриона, поэтому они сказываются на развитии всего организма. Есть несколько внешних признаков, по которым с большой вероятностью можно предположить наличие синдрома Эдвардса у новорожденных.
Одной из наиболее характерных черт этого заболевания является искаженная форма черепных костей: череп вытянут от макушки к подбородку, вместе с тем часто ставится диагноз «микроцефалия» (уменьшение размеров черепа и мозга) или «гидроцефалия» (скопление жидкости в головном мозге).
Лоб узкий, а затылочная сторона более широкая и выступающая, при этом уши расположены ниже, чем при нормальном развитии. Деформируются челюстные кости – часто это приводит к значительному уменьшению нижней челюсти, она становится узкой и недоразвитой. Вследствие этого рот также небольшой и часто треугольный из-за укорочения верхней губы.
Глазные щели уже и короче, чем нужно, переносица расширена и вдавлена – это особенно заметно при том, что нос, как правило, заужен, косточки носа могут визуально отсутствовать. Глазное яблоко также подвержено изменениям и нарушениям, которые приводят к катаракте и колобоме, то есть отсутствию части глазной оболочки. Кроме того, могут быть и иные нарушения зрения.
Уши низко посажены и деформированы, часто в горизонтальной плоскости. На ухе часто отсутствует мочка, а иногда козелок. Наружный слуховой проход часто сужен, иногда может отсутствовать полностью.
Широкий спектр нарушений касается костного скелета. Прежде всего, суставы не могут функционировать нормально, поэтому стопы и кисти не могут сгибаться и разгибаться так, как нужно.
К тому же стопы недоразвиты, из-за этого изменяется их форма, они становятся менее подвижными. Большой палец укорочен, а второй и третий срастаются, иногда настолько сильно, иногда формируются ластообразные конечности.
В 80% случаев формируется стопа с провисающим сводом, выступающей пяткой и коротким большим пальцем.
Из-за излишней подвижности тазобедренного сустава нередко случаются вывихи.
На подушечках пальцев количество дуг может быть в 10 раз больше нормы, однако сгибательной складки пальцы не имеют. Почти у 30% больных на ладонях появляются поперечные борозды и множество гребешков.
Помимо всего прочего, при синдроме Эдвардса деформируется форма грудной клетки – она расширяется, а межреберные промежутки уменьшаются, таким образом она становится короче и шире.
Имеют место очень серьезные нарушения в обменных процессах, таких как работа эндокринной системы. Вследствие хромосомных нарушений железы не могут функционировать нормально, поэтому рост существенно замедляется. Гормональные нарушения приводят к недоразвитости подкожной клетчатки. У каждого десятого отмечается нарушение работы надпочечников или щитовидной железы.
Пониженный мышечный тонус со временем обычно повышается, при этом улучшается кровообращение.
Примерно у половины больных наблюдается аномальное развитие кишечника. Чаще всего эта аномалия заключается в его необычном расположении, при этом появляется мешок, образованный из слоев стенки кишки, а пищевод сужается слишком резко. Почки часто бывают сегментированы или имеют неправильную форму дуги, также может быть удвоение мочеточников.
Изменения также затрагивают и половые органы. У мальчиков яичко может не опускаться в мошонку (крипторхизм) и меняется строение пениса. У девочек формируется гипертрофированный клитор, а яичники развиты недостаточно.
В целом картина внешних и внутренних отклонений при синдроме Эдвардса выглядит следующим образом. В 100% случаев наблюдаются аномалии строения черепа и изменение формы лица.
Почти у 97% уменьшение челюсти (микрогения), чуть более чем в 95 % случаев нарушается строение и расположение ушных раковин.
Удлинение черепа наблюдается почти у 90% пациентов, высокое небо – у 78%, а уменьшенный рот – в 71% случаев.
Развитие сердечно-сосудистой системы нарушено более чем у 90% пациентов. Около 1/3 пациентов имеют нарушения мочеполовой системы, а 55% –пищеварительной системы.
Диагностика
Поскольку синдром Эдвардса характеризуется довольно большим количеством ярко выраженных отклонений, его довольно просто диагностировать даже по внешним проявлениям. Однако этого недостаточно, чтобы поставить окончательный диагноз.
Диагностика синдрома Эдвардса складывается из трех этапов — обследование супружеских пар до момента зачатия, беременной женщины до родов и ребенка после появления на свет.
Диагностика до зачатия ребенка — идеальный вариант, но не всегда применимый. Специалисты-генетики могут лишь предположить, каков риск рождения ребенка с хромосомным заболеванием в данной семье.
До момента зачатия врачи собирают семейный анамнез, опрашивая родителей об их родословной.
Большое внимание специалисты уделяют факторам риска: возрасту матери, перенесенным инфекционным заболеваниям, хроническим болезням, вредным привычкам.
Генетический анализ родителей – полноценное исследование, с помощью которого составляется их кариотип и обнаруживаются участки ДНК с дефектными генами.
Диагностика в период внутриутробного развития дает более точные результаты, поскольку обследуют организм плода. Пренатальная диагностика — важный этап в процессе выявления хромосомных нарушений.
- Ультразвуковое исследование плода и допплерография маточно-плацентарного кровотока – неинвазивные методы, полностью безопасные и рекомендованные всем беременным. Признаки синдрома Эдвардса: отставание плода в размерах и массе, большое количество околоплодных вод, видимые аномалии развития черепа и костей, агенезия пупочной артерии, малая величина плаценты, многоводие, брадикардия, отсутствие носовых костей, 2 артерии в пуповине, кисты сосудистых сплетений. Диагностика с помощью ультразвукового исследования является достоверной на 100%.
- Стандартный пренатальный скрининг включает анализ крови на сывороточные маркеры. Полученные результаты соотносят с возрастом беременной женщины и сроком гестации. При отклонении показателей от нормы ставят высокий риск синдрома Эдвардса. В таких случаях показано искусственное прерывание беременности по медицинским показаниям.
- Амниоцентез – клеточный анализ околоплодных вод. Инвазивная методика, осуществляемая путем забора амниотической жидкости шприцем. Ее клетки содержат образцы ДНК плода, которые проверяют на наличие генетических заболеваний.
- Кордоцентез — исследование пупочной крови плода, позволяющее определить генетические аномалии с высокой точностью.
- Биопсия хориона представляет собой пункцию матки через переднюю брюшную стенку и забор ткани для анализа – стандартного генетического исследования.
Беременным, попадающим в группу высокого риска, предлагается проведение инвазивной дородовой диагностики с последующим кариотипированием плода. Инвазивные методы считаются самыми точными и надежными, но требующими оперативного вмешательства и проникновения в оболочку плода. Диагноз подтверждается при помощи определения кариотипа малыша путем КФ-ПЦР.
Диагностика синдрома Эдвардса после рождения самая легкая, быстрая и точная. После выявления некоторых врожденных дефектов проводят генетический анализ для подтверждения диагноза. Основной задачей при рождении ребенка с этой патологией является обнаружение аномалий в развитии внутренних органов, которые обычно приводят к смерти в первые месяцы жизни. Именно на их поиск направлено большинство диагностических процедур непосредственно после рождения. Наиболее важными диагностическими исследованиями, которые должны быть выполнены ребенку с синдромом Эдвардса в первые часы жизни, являются эхокардиография, УЗИ органов брюшной полости и УЗИ почек.
Причины заболевания
Причиной заболевания является наличие дополнительной 18-й хромосомы (трёх вместо двух в норме для диплоидного набора) в кариотипе зиготы.
Лишняя хромосома обычно появляется до оплодотворения. У человека нормальные половые клетки — гаметы — содержат по 23 хромосомы (гаплоидный набор) и, сливаясь, они дают кариотип зиготы — 46 хромосом. К появлению лишней хромосомы у гамет обычно приводит нерасхождение хромосом при мейотическом делении, вследствие чего в половой клетке оказывается 24 хромосомы. В случае, если такая клетка встретит при оплодотворении гамету от противоположного пола, они образуют зиготу с трисомией.
В одном случае из десяти наблюдается мозаицизм в явлении трисомии 18: лишнюю хромосому несут не все клетки организма. Это говорит о том, что нерасхождение произошло на ранней стадии развития зародыша, а все клетки с трисомией — потомки неправильно поделившейся клетки зародыша.
Проявления синдрома
Дети с трисомией 18 рождаются с низким весом, в среднем около 2200 грамм, при этом длительность беременности — нормальная или даже превышает норму. Фенотипические проявления синдрома Эдвардса многообразны. Чаще всего возникают аномалии мозгового и лицевого черепа, мозговой череп имеет долихоцефалическую форму. Нижняя челюсть и ротовое отверстие маленькие. Глазные щели узкие и короткие. Ушные раковины деформированы и в подавляющем большинстве случаев расположены низко, несколько вытянуты в горизонтальной плоскости. Мочка, а часто и козелок отсутствуют. Наружный слуховой проход сужен, иногда отсутствует. Грудина короткая, из-за чего межреберные промежутки уменьшены и грудная клетка шире и короче нормальной. В 80 % случаев наблюдается аномальное развитие стопы: пятка резко выступает, свод провисает (стопа-качалка), большой палец утолщён и укорочен. Из дефектов внутренних органов наиболее часто отмечаются пороки сердца и крупных сосудов: дефект межжелудочковой перегородки, аплазии одной створки клапанов аорты и лёгочной артерии. У всех больных наблюдаются гипоплазия мозжечка и мозолистого тела, изменения структур олив, выраженная умственная отсталость, снижение мышечного тонуса, переходящее в повышение со спастикой.
Прогноз
Продолжительность жизни детей с синдромом Эдвардса невелика: 60 % детей умирают в возрасте до 3 месяцев, до года доживает лишь 5-10 %. Основной причиной смерти служат остановка дыхания и нарушения работы сердца. Оставшиеся в живых — глубокие олигофрены.
Частота появления
Частота появления синдрома Эдвардса составляет ~ 1:7000 зачатий и 1:8000 рождений живых детей. Риск рождения больного ребёнка увеличивается с возрастом, особенно, если мать болеет диабетом.
Вариации
Кроме трисомии 18, присутствующей во всех клетках организма, а также мозаичной трисомии 18, возможна и частичная трисомия. При этом часть хромосомы 18 присоединяется к другой хромосоме. Такой эффект называется транслокация, и он может произойти как при созревании гамет, так и после оплодотворения в клетках зародыша. В клетках организма при этом оказываются две гомологичные хромосомы 18 и, дополнительно, часть хромосомы 18, прикреплённая к другой хромосоме. У людей, страдающих частичной трисомией 18, аномалии проявляются слабее, нежели при типичном синдроме Эдвардса.
Примечания
- Disease Ontology release 2019-05-13
- Monarch Disease Ontology release 2018-06-29sonu
- Синдром Дауна
- Синдром Патау
- Genetics Home Reference. (англ.). Genetics Home Reference. Дата обращения 20 сентября 2019.
Как выглядит синдром Эдвардса с фото
Современные методы позволяют диагностировать патологию до рождения ребенка. Однако в большинстве случаев заболевание обнаруживается после появления новорождённого на свет. У детей с синдромом Эдвардса присутствуют ярко выраженные аномалии развития. Они дают возможность сразу заподозрить правильный диагноз. Подтверждение выполняется при помощи специального анализа.
Младенцев, родившихся с синдромом Эдвардса, легко отличить от других по внешнему виду. У таких детей присутствуют:
- срощенные пальцы;
- стопа-качалка
- измененная форма черепа;
- аномалии развития половых органов;
- изменение формы нижней челюсти;
- аномалия развития нёба;
- дерматоглифические признаки;
- изменение формы ушных раковин;
- флексорное положение кистей.
Типичным внешним проявлением выступает изменение формы головы новорожденного ребёнка. У детей с таким синдромом имеется более длинный и узкий череп. Аномалия подтверждается при помощи специальных измерений. Другим вариантом аномалии выступает наличие слишком маленькой головы по сравнению с остальным туловищем. Это явление затрагивает именно черепную коробку, в которой размещается мозг.
У всех детей с подобной проблемой присутствуют патологии развития ушных раковин. Этот симптом наблюдается в 95% случаев. Однако при мозаичной форме синдрома Эдвардса шанс на возникновение аномалии снижается. Ушная раковина у таких детей располагается значительно ниже. Иногда она находится ниже уровня глаз. Характерная выпуклость хряща, формирующего ушную раковину, отсутствует. Сам слуховой проход сужен. В 20-25 процентах случаев он может вовсе отсутствовать.
У детей процесс формирования твёрдого нёба во время внутриутробного развития остаётся незавершенным. Там, где у здоровых людей наблюдается срединный шов, у больных заболеванием присутствует продольная щель.
Имеет место быть стопа-качалка. Дефект состоит в неправильном расположении таранной, пяточной и ладьевидных костей. Патологию относят к категории плоского больных деформации стопы у детей. Внешняя стопа новорожденного ребёнка отличается отведением пяточного бугра назад. При этом свод стопы может вовсе отсутствовать.
Больным патологией свойственна ненормальная пропорция длин пальцев. В норме большой палец должен являться самым длинным. У новорождённых с синдромом Эдвардса он уступает по длине второму. По мере роста ребёнка эта особенность становится всё более заметной. Возможно сращение пальцев.
Наблюдается изменение формы нижней челюсти. У больных синдромом Эдвардса подбородок сильно втянут. Это происходит из-за недоразвития нижней челюсти.
Что такое анеуплоидия, трисомия, транслокация, мозаицизм
В каждой клетке человеческого организма находится 46 хромосом, в которых выделяют две группы: 22 пары аутосом (пронумерованных с 1 по 22, в зависимости от размера) и пара половых хромосом (XX у женщин, XY у мужчин). Каждая хромосома в паре является гомологичной другой хромосоме в паре.
В норме человек имеет диплоидный набор хромосом, то есть в каждой клетке содержится двойной комплект каждой из 23 хромосом.
Но есть ситуации, в которых клетки содержат ненормальный, не кратный 46, набор хромосом, что называется анеуплоидией. Анеуплоидия может выражаться, например, в наличии добавочной хромосомы (n + 1, 2n + 1 и т. п.) или в нехватке какой-либо хромосомы (n — 1, 2n — 1 и т. п.).
Формы анеуплоидии:
- моносомия (наличие одной из пары хромосом, например, синдром Шерешевского-Тернера, выражающийся в наличие одной половой Х-хромосомы)
- трисомия (наличие трех вместо 2 хромосом пары).
- тетрасомия (4 гомологичные хромосомы вместо пары в диплоидном наборе)
- пентасомия (5 вместо 2-х) встречаются чрезвычайно редко.
Дальше речь пойдет о самых частых хромосомных аномалиях — трисомиях. В некоторых случаях дополнительная хромосома представлена целой отдельной хромосомой (полная трисомия), а в некоторых этот генетический материал переносится на другую хромосому, что называют транслокацией.
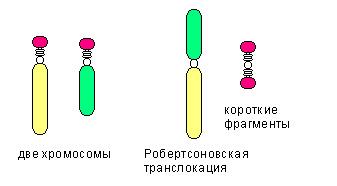
Среди транслокаций также выделяют:
- реципроктную транслокацию, когда неготомологичные хромосомы обмениваются участками
- робертсоновскую транслокацию (см.рис), при которой две неготомологичные хромосомы объединяются в одну.
- Сбалансированная транслокация не сопровождается утратой генетического материала.
Мозаицизмом называют ситуацию, когда среди всех клеток организма есть нормальные, а есть клетки с патологией (например, с трисомией). В этом случае степень отклонений зависит от количества клеток, которые имеет ненормальный генетический материал.
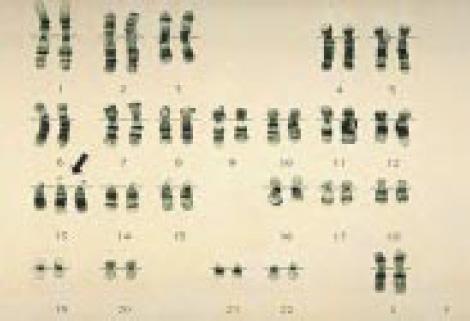
Хромосомы в случае синдрома Патау — Трисомия 13

Хромосомы в случае синдрома Эдвардса — Трисомия 18
Лечение синдрома Эдвардса
Нужно учитывать, что только один из десяти детей, родившихся с подобной патологией, доживают до одного года. Потому первоначальное лечение состоит в избавлении от заболеваний, повышающих риск летального исхода. Если имеет место быть атрезия кишечника или анального прохода, используются меры для дефекации. Ребенка кормят через зонд. Если в организме новорождённого наблюдается активные воспалительные или инфекционные процессы, осуществляется терапевтическое лечение.
Если ребёнка всё же удаётся спасти, в последующем может быть выполнено оперативное вмешательство. В первую очередь выполняется устранение волчьей пасти. Дополнительно проходит борьба с пороками сердца. Пупочная и паховая грыжа удаляются при помощи оперативных методов.
Проводится и медикаментозная терапия. Ребенку назначают лекарства, которые позволяют избавить его от запоров, повышенной кислотности и метеоризма. Родители должны понимать, что синдром Эдвардса способствует появлению следующих патологий:
- отит;
- новообразование в почках;
- гипертензия легких;
- конъюнктивит;
- воспаление лёгких;
- высокое артериальное давление;
- синусит и фронтит;
- инфекционные заболевания мочеполовой системы.
Из-за большого количества патологических изменений, происходящих во внутренних и внешних органах больного, дальнейший прогноз развития неблагоприятен. Те дети, которые сумеют дожить до года и достичь относительно взрослого возраста, будут иметь явное умственное торможение в развитии. Они не смогут самостоятельно ухаживать за собой и удовлетворять свои естественные требования. За такими пациентами необходимо обеспечить постоянный уход и контроль на протяжении всей их жизни.
Однако дети с отклонениями понимают, когда с ними ласково общаются, играют и утешают. Дети с таким синдромом могут научиться самостоятельно есть, улыбаться. Возможно постепенное обучение другим полезным бытовым навыкам.
Высокое количество патологий внутренних органов, развившихся неправильно, приводит к высокому проценту детской смертности. Если в первые месяцы беременности женщина попала в группу риска по этому заболеванию, медики рекомендует выполнить аборт по медицинским показаниям. Однако окончательное решение остается за женщиной.