Невральная амиотрофия шарко–мари–тута
Содержание:
Причины
Генетические заболевания определяются комбинацией генов для конкретного признака, которые находятся на хромосомах, полученных от отца и матери.
Человек получивший один нормальный и один ген заболевания, является носителем, но обычно не проявляет симптомов.
- Риск для двух родителей-носителей передачи дефектного гена детям – 25%.
- Иметь ребенка – носителя -50%.
- Шанс для ребенка получить нормальные гены – 25%.
Риск одинаковый для мужчин и женщин.
Доминантные генетические расстройства возникают, когда для появления болезни необходима только одна копия аномального гена. Аномальный ген может быть унаследован от любого из родителей или быть результатом новой мутации (изменения гена).
Риск передачи ненормального гена от пострадавшего родителя к потомству составляет 50% для каждой беременности независимо от пола ребенка.
Х-сцепленные доминантные генетические нарушения вызваны аномальным геном на Х-хромосоме. Мужчины с аномальным геном страдают более сильно, чем женщины.
Наследственная нейропатия подразделяется на несколько типов, называемых CMT1, CMT2, CMT3, CMT4 и CMTX.
CMT1
Является доминирующей формой расстройства, при котором скорости проводимости нерва являются медленными. Более распространен, чем CMT2. Вызван аномальными генами, которые участвуют в структуре и функции миелина. Дополнительно подразделен на CMT1A, CMT1B, CMT1C, CMT1D, CMT1X на основе специфических аномалий.
- CMT1A появляется из-за дублирования гена PMP22, который расположен на хромосоме 17 при 17p11.2. Является наиболее распространенным типом.
- CMT1B вызван аномалиями в гене MPZ на хромосоме 1 при 1q22.
- CMT1C появляется от аномалий SIMPLE, расположенном на 16 хромосоме при 16p13.1-p12.3.
- CMT1D аномалия EGR2, расположенной на 10, при 10q21.1-q22.1.
- CMT1X возникает от мутаций GJB1 (Xq13.1), Он кодирует белок связного соединения connexin32.
Узнать больше Что такое синдром Шихана?

CMT2
Является аутосомно-доминирующей формой расстройства, при котором скорости проводимости нерва обычно нормальны или немного медленнее, чем обычно. Вызван аномальными генами, участвующими в структуре и функции аксонов. Дополнительно подразделен на CMT2A-2L на основе мутаций.
- CMT2A, является наиболее распространенным и обусловлен ошибками MFN2, расположенным на хромосоме 1, в 1p36.2.
- CMT2B от мутаций RAB7 на хромосоме 3 при 3q21.
- CMT2C вызывается неизвестным геном на 12 – 12q23-34.
- CMT2D ошибки GARS, на 7 – 7p15.
- CMT2E от NEFL, расположенном на 8 – 8p21.
- CMT2F ошибки гена HSPB1.
- CMT2L мутации HSPB8.
Доминирующая промежуточная DI-CMT. Она названа так из-за «промежуточной» скорости проводимости, неопределенности относительно того, является ли нейропатия аксональной или демиелинизирующей. Известно, что доминантные мутации в DMN2 и YARS вызывают этот фенотип.
CMT3
Также называемый болезнью Дежерин-Соттас, у индивидуумов с этим расстройством обнаружена мутация в одном из генов, ответственных за CMT1A, CMT1B, CMT1D, CMT4.
CMT4
Аутосомно-рецессивная форма состояния. Он подразделяется на CMT4A, CMT4B1, CMT4B2, CMT4C, CMT4D, CMT4E, CMT4F.
- CMT4A вызван аномалиями GDAP1. Ген находится на хромосоме 8 при 8q13-q21.
- CMT4B1 – аномалия MTMR2 на 11 – 11q22.
- CMT4B2 от аномалий SBF2 / MTMR13, на 11 при 11p15.
- CMT4C ошибки KIAA1985, на хромосоме 5 – 5q32.
- CMT4D мутации NDRG1, на хромосоме 8 – 8q24.3.
- CMT4E, также известная как врожденная гипомиелиническая невропатия. Происходит от аномалий EGR2, на 10 – 10q21.1-q22.1.
- CMT4F аномалии PRX, на хромосоме 19 – 19q13.1-q13.2.
- CMT4H ошибки FDG4.
- CMT4J мутации FIG4.
Однако большинство случаев CMT2 не вызваны мутациями этих белков, поэтому многие генетические причины еще не обнаружены.
CMTX
Является X-связанной доминирующей формой расстройства. На CMT1X приходится около 90% случаев. Конкретный белок, ответственный за оставшиеся 10% CMTX, еще не идентифицирован.
Аутосомно-рецессивный CMT2 происходит из-за мутаций LMNA, GDAP1.

Структурное выравнивание с элементами вторичной структуры, расположенными сверху. Десять мутаций, вызывающих CMT, отмечены вертикальными стрелками. (Нажмите, чтобы увеличить)
Диагностика
Самый большой риск развития заболевания у лиц, в родстве которых есть люди с подобным заболеванием. Связана патология с поражением периферических нервов. Они, в свою очередь, состоят из аксона и миелиновой оболочки. Если появляется заболевание, то страдать может как оболочка, так и аксон.
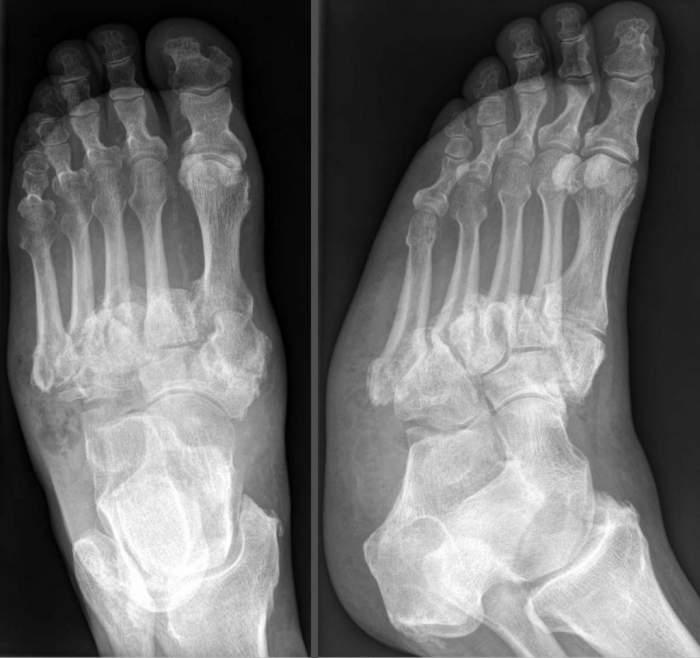
Диагностические мероприятия при подозрении на наличие болезни Шарко начинаются с подробного опроса пациента, выявления наличия родственников с подобным заболеванием. В дальнейшем проводится исследование скорости и силы электрических импульсов, проходящих через нервы. Данное исследование еще называется электромиография. В этом случае в мышцы поочередно вводят тонкую иглу и вызывают слабые поражения электрическим током, если ответ нервной системы слабый или задержанный, то это дает право предположить наличие патологии.
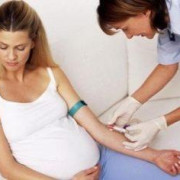
Также обязательно проводится генетическое тестирование для определения наличия или отсутствия мутации гена.
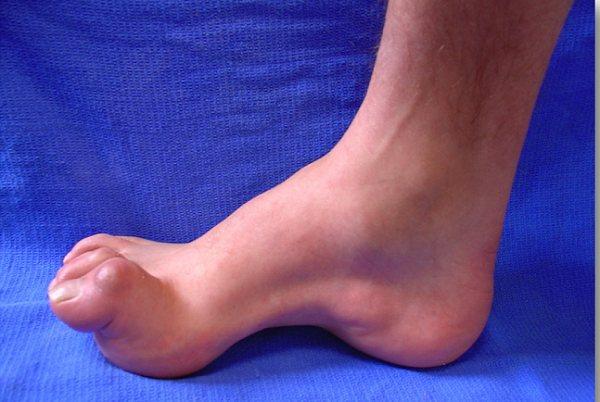
Как проявляется болезнь Шарко-Мари-Тута?
Болезнь Шарко-Мари-Тута проявляется даже в пределах одной семьи не всегда одинаково. И дело не в многообразии её признаков. А в том, что гены, кодирующие данную патологию, способны с разной степенью выраженности формировать симптомы. Проще говоря, имея идентичную «поломку» в хромосомах, признаки недуга у отца и сына будут иметь индивидуальную окраску.
Общие симптомы
Клиническая картина заболевания практически не зависит от его типа и включает:
- атрофию мышц дистальных, то есть наиболее отдалённых от туловища, отделов конечностей;
- снижение сухожильных и периостальных рефлексов;
- изменение чувствительности, характеризующееся её выпадением, но никогда не сопровождающееся появлением ощущения покалывания или «ползания мурашек»;
- деформацию опорно-двигательного аппарата – сколиоз, увеличение свода стопы и др.
Но всё же существует ряд признаков несколько отличающих течение заболевания при различных его вариантах.
Первый тип
Болезнь Шарко-Мари-Тута первого типа нередко протекает в исключительно стёртой форме, при которой пациенты не ощущают изменений в организме и не обращаются за медицинской помощью вообще. Если же патология себя проявила, то происходит это в период первого, максимум второго, десятилетия жизни.
При этом наблюдаются:
- болезненные судороги в мышечном массиве голени, причём редко в икроножной мышце, чаще в передней группе мышц. Подобные спазмы нарастают после периода длительной физической нагрузки (ходьбы, занятий спортом, езде на велосипеде);
- изменения в походке, связанные с постепенным усилением слабости в мышцах. При этом у детей это может дебютировать в хождении на цыпочках;
- деформация стоп с формированием высокого свода последних и наличием молоточкообразных пальцев, которая развивается как результат дисбаланса в тонусе сгибателей и разгибателей;
- атрофия мышц, начинающаяся со стоп и поднимающаяся на голень. Затем процесс затрагивает кисть – появляется тремор в руках и выраженная слабость в пальцах, особенно при попытке выполнять мелкие движения. К примеру, застёгивать пуговицы, перебирать крупу;
- угнетение или полное отсутствие сухожильных и периостальных рефлексов, а именно ахиллового, карпорадиального, при сохранных с более проксимальных отделов рук и ног. То есть коленный и рефлексы с двуглавой и трёхглавой мышцы остаются интактными;
- нарушения чувствительности в кистях и стопах, выражающиеся в её постепенном выпадении. Причём стартует патология с вибрационной и тактильной сферы, распространяясь на суставно-мышечные и болевые ощущения;
- сколиоз и кифосколиоз;
- утолщение нервных стволов, чаще всего поверхностного малоберцового и большого ушного.

Для болезни Шарко характерна атрофия мышц именно в дистальных отделах конечностей. При этом если не выражена подкожно-жировая клетчатка, объём голени и бедра разительно отличается, и ноги приобретают вид таковых у аиста или походят на перевёрнутую бутылку для шампанского.
Невральная амиотрофия Шарко Мари первого типа имеет атипичные формы. Одна из них — синдром Руси-Леви, при котором наблюдается выраженный тремор при попытке удержать руки в одном положении и неустойчивость при ходьбе. Сюда же относится заболевание, проявляющееся, помимо стандартных симптомов, парезами, гипертрофией мышц голени, резким выпадением чувствительности и ночными судорогами в икроножных мышцах.
Второй тип
Для болезни Шарко-Мари-Тута второго типа, кроме более позднего начала, характерны:
- менее выраженные изменения чувствительности;
- более редкая встречаемость деформаций стопы и пальцев;
- наличие синдрома беспокойных ног (возникают неприятные ощущения в ногах во время отхода ко сну, заставляющие пациента двигаться, что облегчает состояние);
- нередко сохранная сила в кисти;
- отсутствие утолщения нервных стволов.
При синдроме Шарко-Мари-Тута, передающемся через Х-хромосому, могут встречаться нейросенсорная тугоухость (снижение слуха) и транзиторная энцефалопатия, возникающая после физической нагрузки на высоте. Для последней характерно появление симптомов через 2-3 дня после занятий. Признаками патологии становятся шаткость, нарушение речи, глотания, слабость в проксимальных отделах рук и ног. Обычно клиническая картина недуга исчезает самостоятельно в течение пары недель.
Болезнь Шарко симптомы
Одним из самых важных клинических симптомов начальной стадии Болезни Шарко является атрофия мышц с ассиметричным прогрессированием. Заболевание может начаться с любой группы мышц, впоследствии поражение становится генерализованным. Выделяют несколько видов параличей. К ним относят шейно-грудной, псевдобульбарный, бульбарный и пояснично-крестцовый. Летальный исход наступает через несколько лет, после вовлечения в прогрессирующий процесс мышц респираторного характера.
Очень часто, почти в 40% случаев, характерной картиной болезни Шарко является слабость мышц какой-то одной из верхних конечностей. Поражение начинается с кисти, за которой следует быстрое прогрессирование заболевания. Больной испытывает затруднения в работе указательного и большого пальцев, также нарушается тонкий моторный контроль. Это проявляется в затруднительном подбирании предметов во время одевания, особенно, при застёгивании пуговиц. Во время поражения основной руки, отмечаются проблемы при письме, а также в повседневной жизни.
Для типичного протекания болезни Шарко характерно неуклонное вовлечение остальных мышц этой же конечности, а затем поражение переходит на другую руку. Это предшествует парализации нижних конечностей и бульбарных мышц.
Иногда болезнь Шарко начинает развиваться с мимических мышц, затрагивает язык или, наоборот, мышцы туловища. Поэтому вовлекая новые мышцы в прогрессирующий процесс, болезнь характеризуется короткой продолжительностью жизни человека. Особенно это касается бульбарной формы, при которой пациенты не доживают до паралича ног. Относительно благоприятной считается пояснично-крестцовая форма. Бульбарный и псевдобульбарный паралич проявляется симптомами дизартрии и дисфагии, за которыми наступают респираторные нарушения.
Для всех форм болезни Шарко характерен повышенный нижнечелюстной рефлекс. Дисфагия чаще наблюдается после глотания жидкой пищи, чем после твёрдой. Постепенно ослабевают жевательные мышцы, происходит свисание мягкого нёба и атрофируется язык. Больные с такими симптомами не могут глотать, произносить звуки, у них отмечаются генерализованные судороги.
Атрофия мышц имеет избирательное поражение. Например, на руках – это мышцы у основания большого пальца и указательного, межкостные и дельтовидные; на ногах – только те мышцы, которые осуществляют сгибание с тыльной стороны стопы; бульбарные – мышцы нёба и языка.
Очень редкими клиническими симптомами болезни Шарко считаются поражения глазных мышц. Кроме того, у больных с боковым амитрофическом склерозом, даже обездвиженных, отсутствуют пролежни, что является особенным признаком этого заболевания. Также описаны случаи, когда в процесс парализации равномерно вовлекаются мотойнеры как верхние, так и нижние, при которых преобладают определённые пирамидные или переднероговые синдромы.
Процедуры и операции
Особое внимание уделяется немедикаментозным методам терапии. Комплекс мероприятий, позволяющих достичь максимального терапевтического эффекта:
- Лечебная физическая культура. Регулярные занятия ЛФК повышают мышечный тонус. Наибольший эффект достигается при достижении пассивных упражнений (занятия вместе со специалистом) и активных (выполняются самостоятельно).
- Электростимуляция. Направленная подача электрических импульсов улучшает проводимость по периферической нервной системе, активирует метаболические процессы в паретичных мышцах, усиливает нейротрофику.
- Бальнеотерапия. Грязевые аппликации и грязевые ванны замедляют формирование контрактур и улучшают работу вегетативной нервной системы.
- Массаж. Различные виды массажа (аппаратный и ручной) улучшают лимфатический отток и нормализуют кровообращение. Рекомендован расслабляющий, стимулирующий и вибрационный массаж.
- Ортопедическая терапия. Ношение специальной ортопедической обуви позволяет предотвратить развитие грубой деформации. Из-за мышечной слабости развивается нестабильность суставов. Подтяжки, ортезы и другие специальные приспособления используются для фиксации стоп в необходимом положении.
При комплексном подходе к терапии повышается уровень мышечной силы, нормализуется походка и исправляются различные нарушения равновесия. Грамотно подобранное лечение повышает социальную и бытовую адаптацию, восстанавливает работоспособность пациента.
Лечение амиотрофии
Лечение амиотрофии заключается в остановке атрофии мышечной системы, а в некоторых случаях возможно даже рост новых мышц. Но сразу надо предупредить всех родных больного, что в лечении они тоже должны принять самое непосредственное участие. Ведь лечебные процедуры проводятся не только в больнице, но и дома. И их надо проводить ежедневно, желательно без перерывов, иначе все затраченные усилия пойдут прахом.
Вот примерная схема лечения:
- Прием лекарственных средств по определенной схеме, сроком до двенадцати месяцев
- Специальное питание
- Физиотерапия и массаж
- Лечебная гимнастика
- Психологическая помощь больному
- Для детей, которые из-за болезни отстают в психологическом развитии, оказывается нейропсихологическая помощь.
Лечение лекарственными препаратами делится на три взаимосвязанных направления. Первое из них — это улучшение питания мышц больного. В нем участвуют витамины, аминокислоты, биостимуляторы. Следующим направлением идут те препараты, которые способны стимулировать рост мышечной массы. Здесь задействованы анаболики. И третье направление — это защита мышечных клеток антиоксидантами. Только так взаимодействием нескольких групп препаратов можно бороться с амиотрофией. Дозы, сочетания лекарственных средств подбираются индивидуально для каждого больного. Это зависит от возраста, пола и наличия сопутствующих болезней.
Питание также играет важную роль в лечении таких больных, ведь болезнь ведет к потере мышечной массы больным. Поэтому при помощи питания надо эту потерю восполнить. В этом поможет белковое питание. Идеально для этого подходят питательные смеси — аминокислоты, в которые добавлены витамины и L-карнитин. Но иногда некоторые сопутствующие болезни воспрепятствуют усвоению белков. Тогда в первую очередь лечится причина, из-за которой белок не принимается организмом больного.
Физиотерапия и массаж необходимы для улучшения кровоснабжения ослабленных мышц. Например, физиотерапия убирать из мышечной ткани жировую и рубцовую ткань. Они образуются на месте погибших мышц. Физиотерапия также помогает при контрактурах — это когда снижается амплитуда движений суставов. При применении массажа надо учитывать специфику болезни, ведь стандартные приемы здесь не подходят. При амиотрофии упор при массаже делается на ослабленные, неэластичные участки мышц организма.
При проведении гимнастических упражнений надо учитывать особенности человеческого организма. Дело в том, что у человека при отказе одной групп мышц в работу включаются другие, которые берут на себя функции больного участка. Это может привести к неправильному ходу лечения. Поэтому при проведении гимнастических упражнений надо подбирать те, которые могут изолированно отрабатывать ослабленные мышцы.
Список источников
- Дадали Е.Л., Тибуркова Т.Б., Щагина О.А., Поляков А.В. «Алгоритм диагностики наследственных моторно-сенсорных невропатий», статья из журнала «Нервные болезни», 2010
- Муртазина А.Ф., Щагина О.А, Никитин С.С., Дадали Е.Л. , Поляков А.В. «Современные клинико-генетические представления об аутосомно-рецессивных наследственных периферических нейропатиях», статья в журнале «Анналы клинической и экспериментальной неврологии» №1, 2019
- Каук О.И. «Наследственные полиневропатии как фактор статокинетических нарушений у детей раннего возраста» статья в Международном медицинском журнале №1, 2016
Лечение
Основная причина развития невральной амиотрофии является генетическая предрасположенность. Вылечить полностью заболевание не получится, но замедлить прогрессирование патологии можно. Существует ряд методов, которые помогают устранить симптоматику болезни.
Применяются следующие способы терапии:
- Физиотерапевтические процедуры;
- Лечебные физические упражнения;
- Лекарственные средства;
- Массаж;
- Хирургические вмешательство.=
Если соблюдать все рекомендации врача, то это поможет устранить симптомы в короткие сроки. Главное, вовремя обратиться к врачу для того чтобы он назначил лечение. Если игнорировать первые проявления невральной амиотрофии то это может привести к серьезным последствиям.

Назначаются следующие медикаменты для облегчения состояния больного:
- курс специальных витаминных добавок, чтобы улучшить питание мышечных волокон;
- Нивалин и Прозерин помогает ускорить нервную проводимость;
- Никотиновая кислота, Галидор приводят в норму кровоснабжение.

Лечение подбирается под каждого пациента индивидуально. Всё зависит от того на какой стадии развития заболевание. Для того чтобы больной почувствовал себя легче, нужно будет применять определенные методы лечения. Как правило, одних медикаментов бывает недостаточно, поэтому необходимо сочетать с другими способами терапии. Лучше всего обращаться к врачу при первых проявлений патологии.
Массажные процедуры хорошо помогают справиться с признаками невральной амиотрофии. Массаж делается, как руками, так и аппаратом и происходит воздействие на тело. Массажные процедуры захватывают кожу, мышцы, сосуды, нервы. Если использовать несколько видов массажей, то это прежде всего поможет расслабиться. Можно добиться тонизирующего эффекта, болеутоляющего и так далее.
Важно также соблюдать правильное питание, ведь это играет большую роль в терапии. Невральная амиотрофия приводит к утрате мышечной массе, поэтому придется, чтобы рацион был сбалансированным
Необходимо, чтобы была белковая диета и в питании присутствовали аминокислоты. Может быть так, что организм не усваивает белок. Тогда придется искать причину почему, так происходит.
Должны обязательно проводится физические упражнения. Перед тем, как их выполнять врач должен индивидуально подбирать. При заболевании происходит отказ определенных мышц и другие берут на себя работоспособность пораженных участков. Если неправильно будут назначены упражнения, то от лечения не будет никакого эффекта. Только врач должен подбирать под каждого больного определенные физические нагрузки.
Взрослым людям часто нужна психологическая помощь при заболевании. Когда происходит развитие патологии, то у них могут возникать нехорошие мысли. На подобии того, что они приносят только лишние проблемы своим родным из-за болезни.

В детском возрасте нужна помощь только в том, случае, если необходимо развиваться интеллектуально. Ведь, как правило, мышцы также учувствуют в развитии головного мозга. Для того чтобы не допустить отставание ребенка нужно применять специальные интеллектуальные упражнения.
Существует ряд продуктов, которые запрещается употреблять при заболевании. Отказаться необходимо от алкоголя и энергетиков. Сладкие напитки и кофе также нужно исключить из рациона. Употребление соли необходимо сократить, так как это провоцирует развитие заболевания. Продукты быстрого приготовления и фаст-фуд приводят к разрушению мышечных волокон. Патология называется Шарко-Мари-Тута или наследственная моторно-сенсорная нейропатия.
Список литературы
- МКБ-10 (Международная классификация болезней)
- Юсуповская больница
- Батуева Е.А., Кайгородова Н.Б., Каракулова Ю.В. Влияние нейротрофической терапии не нейропатическую боль и психовегетативный статус больных диабетической нейропатией // Российский журнал боли. 2011. № 2. С. 46.
- Бойко А.Н., Батышева Т.Т., Костенко Е.В., Пивоварчик Е.М., Ганжула П.А., Исмаилов А.М., Лисинкер Л.Н., Хозова А.А., Отческая О.В., Камчатнов П.Р. Нейродикловит: возможность применения у пациентов с болью в спине // Фарматека. 2010. № 7. С. 63–68.
- Морозова О.Г. Полинейропатии в соматической практике // Внутренняя медицина. 2007. № 4 (4). С. 37–39.
Лечение болезни Шарко-Мари-Тута
(с) The New York Times / Michael Nagle
Пока нет никакого лечения для ШМТ, но можно облегчить симптомы и отсрочить начало инвалидности.
НПВС (нестероидные противовоспалительные препараты), такие как ибупрофен, уменьшают суставные и мышечные боли, а также боли, вызванные поврежденными нервами.
Трициклические антидепрессанты (ТЦА) назначают, если НПВС не эффективны. ТЦА обычно используют для лечения депрессии, но они могут уменьшить болевые симптомы невропатии. Тем не менее, они имеют побочные эффекты.
Физическая терапия поможет укрепить и растянуть мышцы. Упражнения помогут сохранить мышечную силу.
Трудотерапия может помочь пациентам, которые имеют проблемы с движениями пальцев и им трудно осуществлять повседневную деятельность.
Ортопедические устройства могут предотвратить травмы. Обувь с высокими голенищами или специальные ботинки обеспечивают дополнительную поддержку голеностопного сустава, и специальная обувь или стельки для обуви могут улучшить походку.
Операция по удалению ахиллова сухожилия иногда может облегчить боль и сделать ходьбу легче. Хирургия может исправить плоскостопие, облегчить боль в суставах.
Осложнения
Если запустить невральную амиотрофию, то могут быть необратимые последствия. Проявляются они в виде нарушения дыхательной системы. Если патология поражает нервные окончания, которые контролируют диафрагму. Больному скорее всего нужно будет употреблять бронхолитические средства или искусственную вентиляцию легких. Лишний вес или сильное ожирение может привести к тому, что пациенту становится трудно дышать.
Депрессивное состояние может быть из-за стрессовых ситуаций. Чаще всего возникает это из-за прогрессирования патологии. Могут применяться, как специальные лекарственные препараты, так и психологическая помощь. Если заболевание будет в запущенной форме и без лечения, то это приведет к инвалидности.

Пациент может перестать полностью передвигаться и на этой фоне возникнуть глухота. Для того чтобы не было такие серьезных последствий, необходимо своевременно обращаться к врачу. Он сможет назначить комплексную диагностику, чтобы поставить точный диагноз. Только после этого будут применяться методы лечения. Заболевание предоставляет хроническую моторную и сенсорную полинейропатию.
Лечение болезни Шарко-Мари-Тута
Лечение назначается только после подтверждения диагноза врачом-специалистом. Показаны дозированная ЛФК и массаж, ортопедические мероприятия, витаминные препараты, средства нейротрофического действия, улучшающие микроциркуляцию, антихолинэстеразные препараты.
Основные лекарственные препараты
Имеются противопоказания. Необходима консультация специалиста.



- (средство, улучшающее метаболизм и энергообеспечение тканей). Режим дозирования: в/м, в первые 2-3 дня вводят 1 раз в день по 1 мл 1%-ного раствора, в последующие дни 2 раза в день или сразу 2 мл 1%-ного раствора 1 раз в день. На курс лечения — 30-40 инъекций.
- (средство, улучшающее микроциркуляцию). Режим дозирования: внутрь, проглатывая целиком, во время или сразу после приема пищи, запивая достаточным количеством воды, в дозе 100 мг 3 раза в сутки с последующим медленным повышением дозы до 200 мг 2-3 раза в сутки.
- (комплекс витаминов группы В). Режим дозирования: терапию начинают с 2 мл внутримышечно 1 р/д на протяжении 5-10 дней. Поддерживающая терапия — 2 мл в/м два или три раза в неделю.
- (анаболическое стероидное средство). Режим дозирования: внутрь, перед едой в дозе 0,005-0,01 г 1-2 раза в день. Курс лечения у взрослых длится 4-8 недель. Перерывы между курсами 4-8 недель.
- (ноотропное средство). Режим дозирования: применяют парентерально в виде в/м инъекций (до 5 мл) и в/в инъекций (до 10 мл). Препарат в дозе от 10 мл до 50 мл рекомендуется вводить только посредством медленных в/в инфузий после разведения стандартными растворами для инфузий. Продолжительность инфузий составляет от 15 до 60 мин. Вводят парентерально в дозе от 5 мл до 30 мл/сут. Рекомендуемый оптималь-ный курс лечения — ежедневные инъекции в течение 10-20 дней.
- (антихолинэстеразное средство). Режим дозирования: внутрь, суточная доза для взрослых составляет 10-40 мг в 2-4 приема.
Наследственное заболевание. Основной тип передачи – аутосомно-доминантный (с пенетрантностью патологического гена около 83%), реже – аутосомно-рецессивный.
Морфологическую основу болезни составляют дегенеративные изменения главным образом в периферических нервах и нервных корешках, касающиеся как осевых цилиндров, так и миелиновой оболочки. Иногда наблюдаются гипертрофические явления в интерстициальной ткани. Изменения в мышцах носят преимущественно неврогенный характер, отмечается атрофия отдельных групп мышечных волокон; в неатрофированных мышечных волокнах структурные изменения отсутствуют. По мере прогрессирования заболевания появляются гиперплазия интерстициальной соединительной ткани, изменения в мышечных волокнах – их гиалинизация, центральное смещение ядер сарколеммы, гипертрофия некоторых волокон. В более поздних стадиях болезни отмечаются гиалиновая дегенерация, распад мышечных волокон. Наряду с этим в ряде случаев отмечены изменения в спинном мозге. Они складываются из атрофии клеток передних рогов, главным образом в поясничной и шейной части спинного мозга, и различной степени поражения проводниковых систем, характерного для наследственной атаксии Фридрейха.